
又是一篇准备了很久的文章。来跟大家分享一下怎样发现并命名一个 SARS-Cov-2 的新毒株。
众所周知,SARS-Cov-2 是 RNA 病毒,容易变异,特别在大规模爆发的情况下容易产生大量的变种。而有一批人时刻关注着全世界上传的病毒测序数据,试图从中整理和分析出新的变种,并总结病毒的演化规律,给与新变种合适的命名。而做这件事的不仅有学术界的相关研究者,许多业余爱好者也能参与,甚至是在座的你在阅读完这篇文章后,或许也能独立完成这个工作(希望如此
前置准备
首先你需要一个 GISAID 账号。GISAID 是全球最大的流感病毒基因组数据共享平台,基本上世界各地的研究机构都会向 GISAID 上传 SARS-Cov-2 的测序数据。GISAID 是一个半封闭的平台,你需要申请一个账号才能访问其数据,并且根据其用户协议不允许将数据分享到平台外。
你需要一个学校或学术机构邮箱以申请 GISAID 账号,否则通过率较低。
尽管还有 GenBank 这种更开放的数据平台,但大部分研究机构更倾向于向 GISAID 上传数据。因此 GISAID 的数据会更及时、更全面。
前置知识
病毒结构
SARS-Cov-2 是一种 RNA 病毒,RNA 即核糖核酸,由核糖核苷酸组成,核苷中起配对作用的部分成为碱基。RNA 中最基本的四种碱基为腺嘌呤(A)、尿嘧啶(U)、鸟嘌呤(G)、胞嘧啶(C)。通常我们也用 AUGC 这四个字母的序列来表示 RNA 的序列。
例如,在常用的表示核酸序列的 fasta 文件中,一个 SARS-Cov-2 病毒可能是这样的:
>hCoV-19/Sichuan/MSCDC-23006/2023|EPI_ISL_17309083|2023-03-01
GATCTGTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTA
ATAACTAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCA
GCCGATCATCAGCACATCTAGGTTTTGTCCGGGTGTGACCGAAAGGTAAGATGGAGAGCCTTGTCCCTGGTTTCAACGAG
AAAACACACGTCCAACTCAGTTTGCCTGTTTTACAGGTTCGCGACGTGCTCGTACGTGGCTTTGGAGACTCCGTGGAGGA
GGTCTTATCAGAGGCACGTCAACATCTTAAAGATGGCACTTGTGGCTTAGTAGAAGTTGAAAAAGGCGTTTTGCCTCAAC
TTGAACAGCCCTATGTGTTCATCAAACGTTCGGATGCTCGAACTGCACCTCATGGTCATGTTATGGTTGAGCTGGTAGCA
GAACTCGAAGGCATTCAGTACGGTCGTAGTGGTGAGACACTTGGTGTCCTTGTCCCTCATGTGGGCGAAATACCAGTGGC
TTACCGCAAGGTTCTTCTTCGTAAGAACGGTAATAAAGGAGCTGGTGGCCATAGGTACGGCGCCGATCTAAAGTCATTTG
ACTTAGGCGACGAGCTTGGCACTGATCCTTATGAAGATTTTCAAGAAAACTGGAACACTAAACATAGCAGTGGTGTTACC
.....
(SARS-Cov-2 病毒大概有三万个碱基,此处省略..)
生物好的孩子可能要问了,RNA 里哪来的 T?是的,众所周知 RNA 里是没有胸腺嘧啶(T)的,但是 fasta 文件约定无论是 DNA 还是 RNA,U/T 都统一用 T 表示。
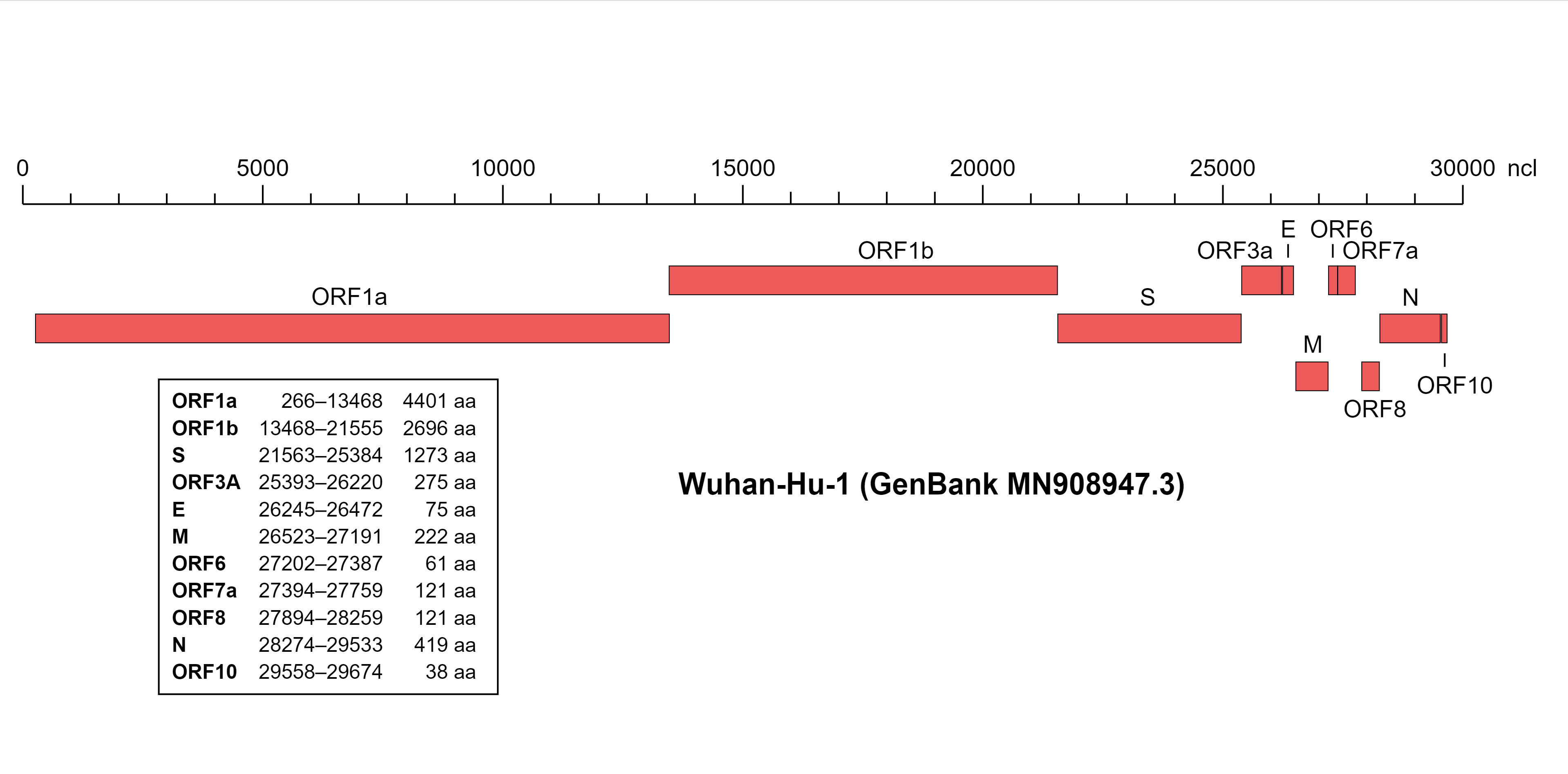
SARS-Cov-2 的序列根据其功能可以分为多个区域,以武汉原始株为例(为例,指正好有个图,实际上所有毒株都有这些区域),如下图,分为 ORF1a、ORF1b、S、ORF3A、E、M、ORF6、ORF7a、ORF8、N、ORF10 共 11 个区域。每个区域的作用就不展开讲了,有兴趣的同学可以自行 Google。
特别的,有一个称为 RBD 域的部分。受体结合域(Receptor-Binding Domain, RBD)是 SARS-Cov-2 病毒上的关键部分,位于病毒的 S(Spike)区域第 318 个到第 541 个氨基酸(可能有多种说法,但反正就这附近),使其能够结合细胞受体(SARS-Cov-2 病毒结合 hACE2 受体)、进入细胞并导致感染。
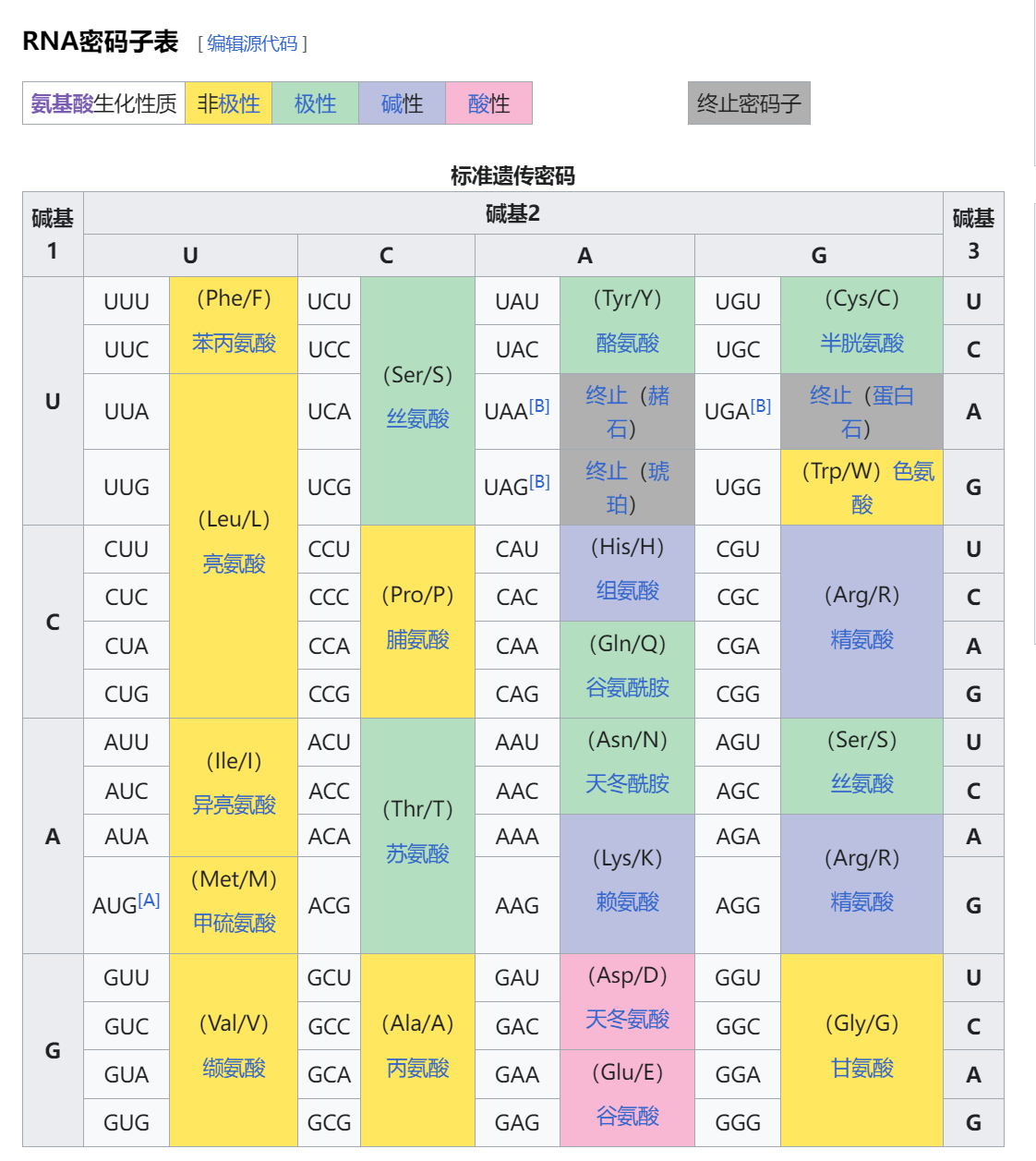
这里图上的 AA 指的是碱基序列对应的氨基酸,众所周知三个碱基可以对应一个氨基酸,来复习一下吧。
突变表示和变异株命名
表示病毒基因序列变化一般有两个维度,碱基维度和氨基酸维度。
碱基维度,比如说病毒的第 12345 位碱基从 A 突变成了 T,就表示为 A12345T。
氨基酸维度,比如病毒 S 蛋白的第 182 个氨基酸,由于碱基突变从赖氨酸变成了谷氨酰胺,就表示为 S:K182Q。
是不是非常简单易懂。注意这里所有的突变的表示都是相对于参考毒株,也就是最早从武汉采集到的那一个,编号 Wuhan-Hu-1 (GenBank:MN908947.3)。
而变异株的命名则要稍微复杂一些,目前普遍采用的命名法是 PANGO 命名法。
首先将 SARS-Cov-2 的最初的两种毒株命名为 A 和 B,而其他所有毒株均是由这两种演化而来。
例如 Omicron 变种的 PANGO 编号为 B.1.1.529,其含义为 B 的第 1 种子毒株的第 1 种子毒株的第 529 种子毒株。
相对于某种毒株的子毒株就在该毒株后增加一节编号,例如在 B.1.1.529 的基础上新增了 S:L452R 和 S:F486V 突变的某种毒株,在命名时应该分配编号 B.1.1.529.X,此处 X 取决于之前已经有多少个基于 B.1.1.529 的子毒株。在该毒株命名时已经有 4 个兄弟毒株,因此它被命名为 B.1.1.529.5。
由于无限增加编号小节会让名称过于冗长,所以规定当小节数超过 3 时,就要分配一个别名编号,别名编号从 C 起编。当 26 个字母用尽时,开始使用两个字母作为编号,例如 AA, AB, …
B.1.1.529.5 最终被命名为 BA.5。
特殊的,重组毒株会以 X 开头,从 XA 起编,26 个字幕用完后用两位字母,例如 XAA, XAB, …
关于命名规则的完整描述,请参见:https://www.pango.network/the-pango-nomenclature-system/statement-of-nomenclature-rules/
实战一下
终于讲完最小限度的基础知识了,因为我也并非专业人士所以一切不需要外延的点全部都略过了,只讲了最最主要的部分。如果你想深究其原理,可能就得自己找点资料看看了~
这里用前几天上海等地上传的序列数据,做一个演示。
首先我们要对序列进行分型。目前主要的分型用的工具有 pangoLEARN 和 nextclade。这里用 nextclade 因为它可以对齐序列分段,而且有一个非常好用的 WebUI。
将 fasta 文件输入,等待分析。结果大概是这样:
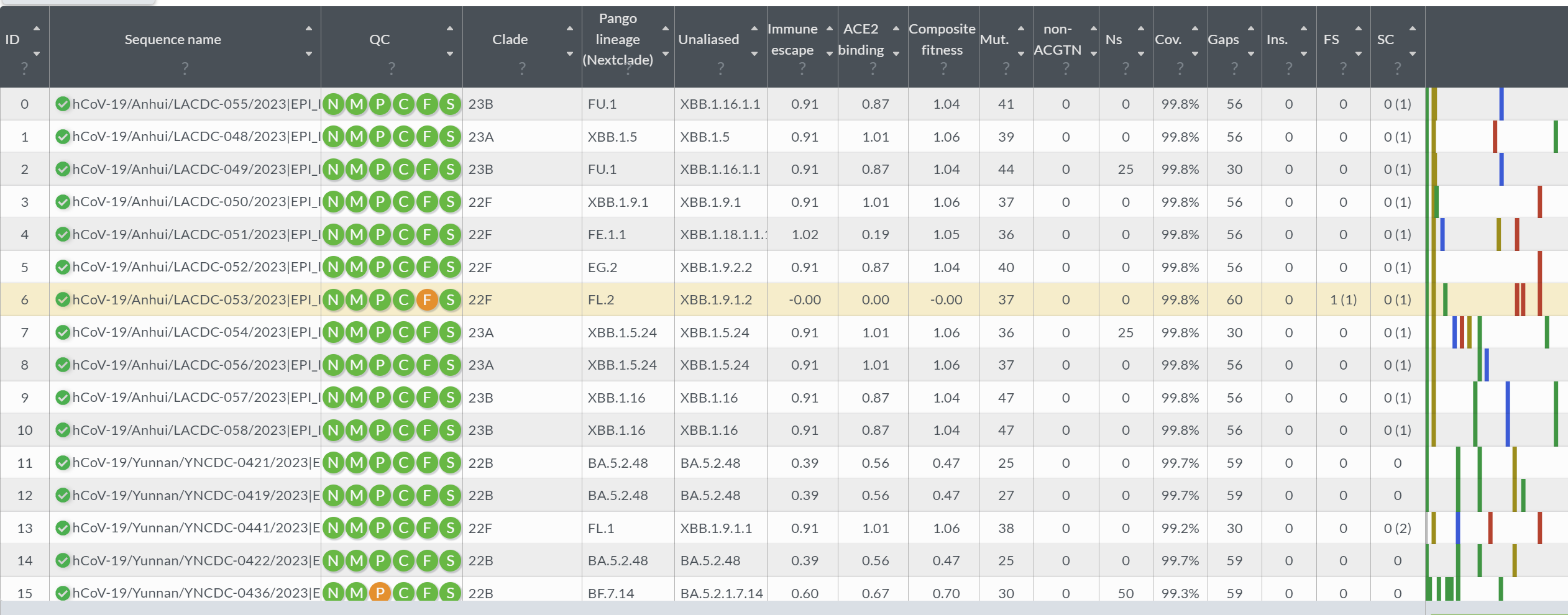
最右侧的竖条标注的就是相对于参考株的突变位点,例如:
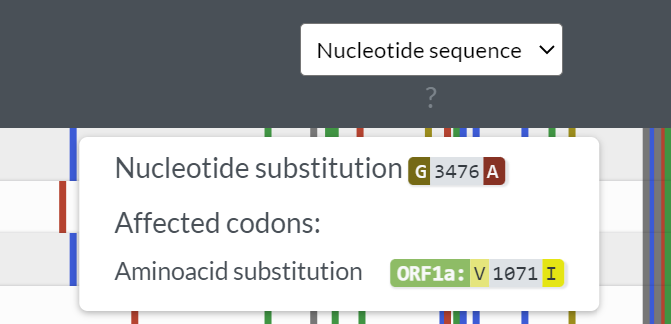
这个就表示 3476 位碱基 G 突变为 A,同时这也导致 ORF1a 区域的第 1071 个氨基酸由 V 变为 I。
所以,我们可以关注某一分型中部分毒株都含有的某个突变位点,这可能预示着一种新毒株。
经过一番搜索,我们发现这一组数据中,部分 DY.1 毒株都具有 G23426A(S:V622I) 和 C24797T(S:P1079S) 这两个突变。
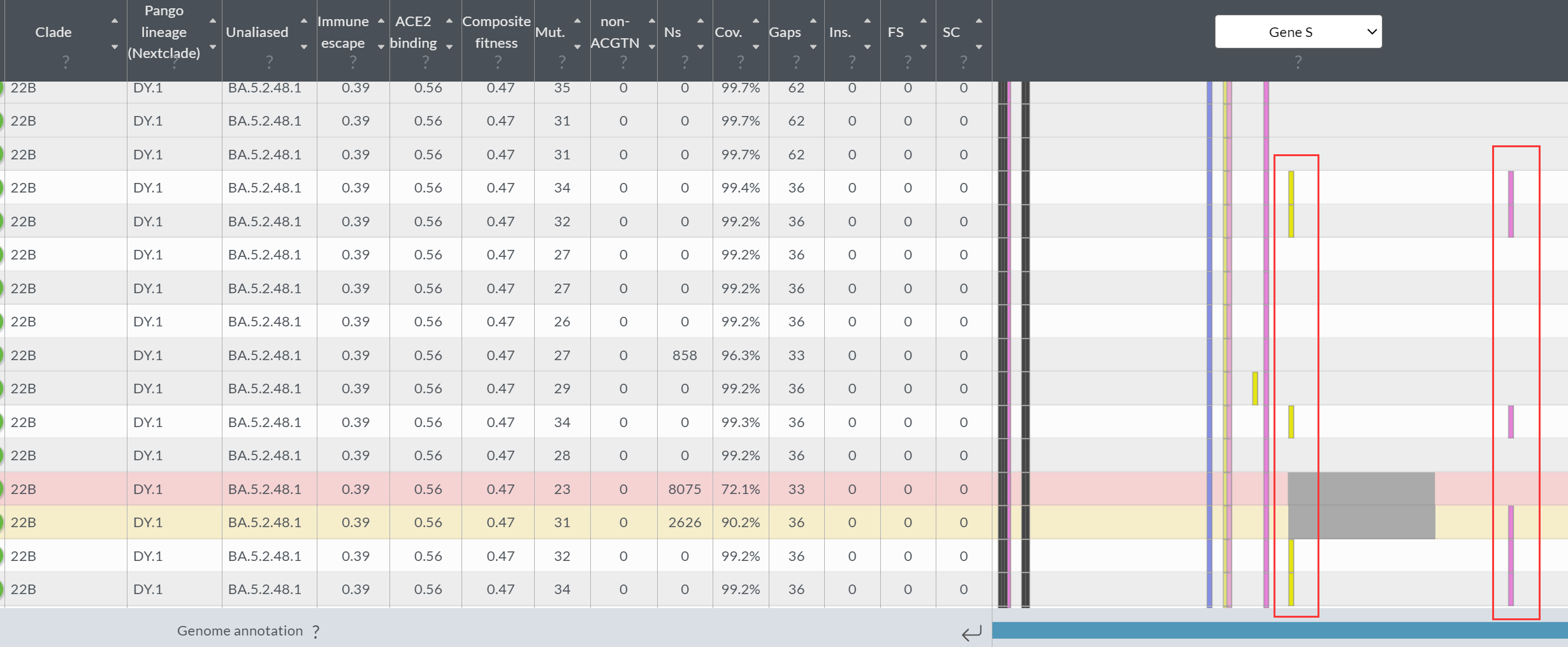
查阅当前所有变异株列表得知,这两个突变并非 DY.1 的定义突变,因此,这可能确实是一小撮新的毒株。
在初步简历目标之后,我们需要将具有这个突变的毒株筛选出来。这里我们可以使用 GISAID 的 AA Substitutions 筛选或者 Nucl Mutations 筛选。经过一番试探,这里选择了使用 BA.5.2.48 + Spike_A570S, Spike_V622I, Spike_P1079S 的条件搜索。其中 BA.5.2.48 + Spike_A570S 可以筛选出所有 DY.1 毒株(DY.1 的定义突变也可以在列表中查到),后面两个突变则是筛选出本次发现的毒株。
结果是搜出来了 14 条结果,把他们的 fasta 下载过来,重新塞进 Nextclade Web 确保他们都是同一种毒株。此外,我们还需要画出这一批毒株在演化树上的位置,这里可以用 UShER 这个工具。输入的 ID 可以用 GISAID 里全选 → 右下角 Select 来生成。UShER 的数据稍有延后,如果是最新的采样需要过两天再试试。
UShER 最后输出的结果放在这里:https://nextstrain.org/fetch/genome.ucsc.edu/trash/ct/subtreeAuspice1_genome_2637b_b973a0.json?f_userOrOld=uploaded sample
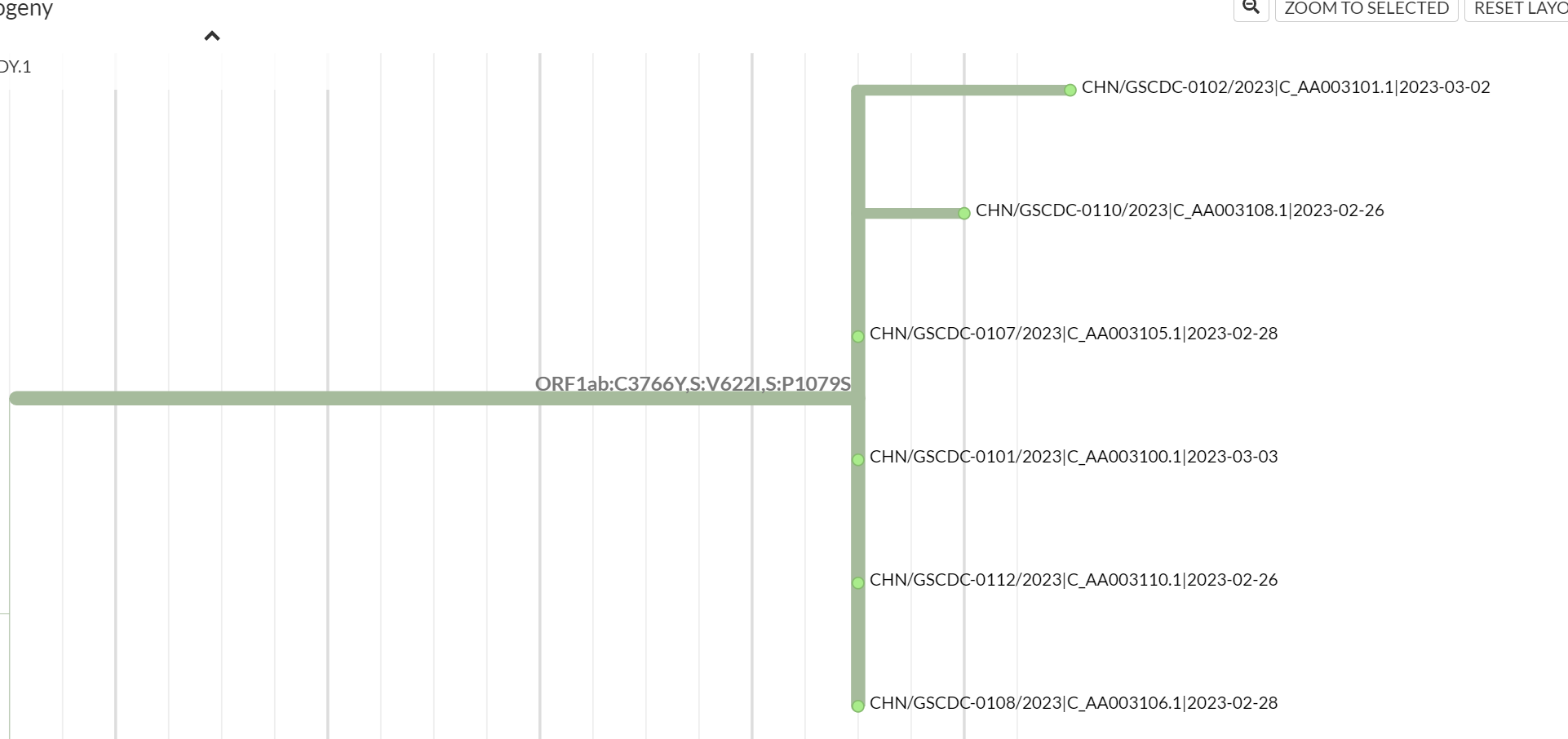
可以看到确实是在同一演化分支上。由于 UShER 数据延后,有一些毒株缺了,问题不大。
这里 UShER 还发现这一批毒株同时都有 ORF1a:C3766Y 这个突变,刚刚我们只关注了 Spike 区域,漏掉了这个。
那么我们就初步描述出了这一组毒株,它可以表示为 DY.1 + S:V622I + S:P1079S + ORF1a:C3766Y。
下一步,我们可以向 PANGO 委员会提交命名申请,如果合适他们会分配一个新的编号给这一组毒株。在写这篇文章的时候,按照命名规则,应该命名为 DY.1.2 (B.1.1.529.5.2.48.1.1.2)。
提交命名的方法就是在 GitHub 的这个仓库开一个 Issue,按照模板填写上面我们获得的信息。
但是!这一毒株很可能并不会得到命名。
为什么呢?
因为每天世界各地上传的测序数量很多,里面含有的新变异株也是茫茫多的,如果每一种毒株都进行命名,显然浪费人力。能够得到命名的毒株总得有其过人之处,比如
- 在当地获得传播优势或者已经输出多个地区。 本次发现的这一撮,全部来自甘肃省的采样,分布在3月-4月发生,共14例,最多只能叫零星发生。关于传播优势,可以使用 covSPECTRUM 这个网站计算,用法这里就不展开了。
- 发生跳跃突变,从上级节点突然获得了好多突变。
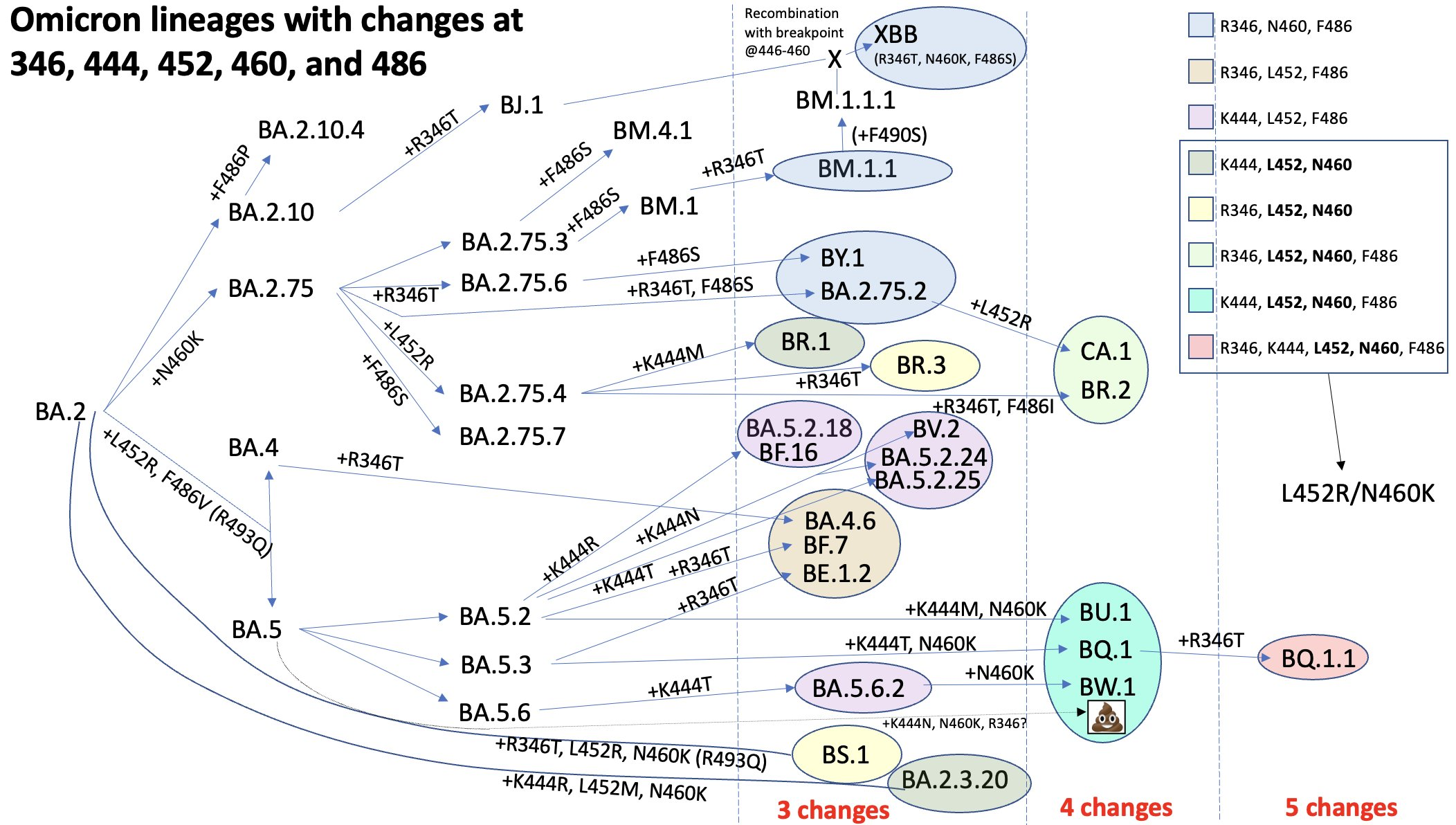
- 趋同演化,获得了当前世界范围内流行毒株的同款突变。 趋同演化是 Omicron 时期病毒演化的一大趋势,指的是世界范围内的多种流行毒株同时独立获得相同位点的突变,例如 Spike 上的 486, 444, 346 等位置[1][2]。有的突变已经证明对传播力有正向效果。因此这些突变会被重点关注。
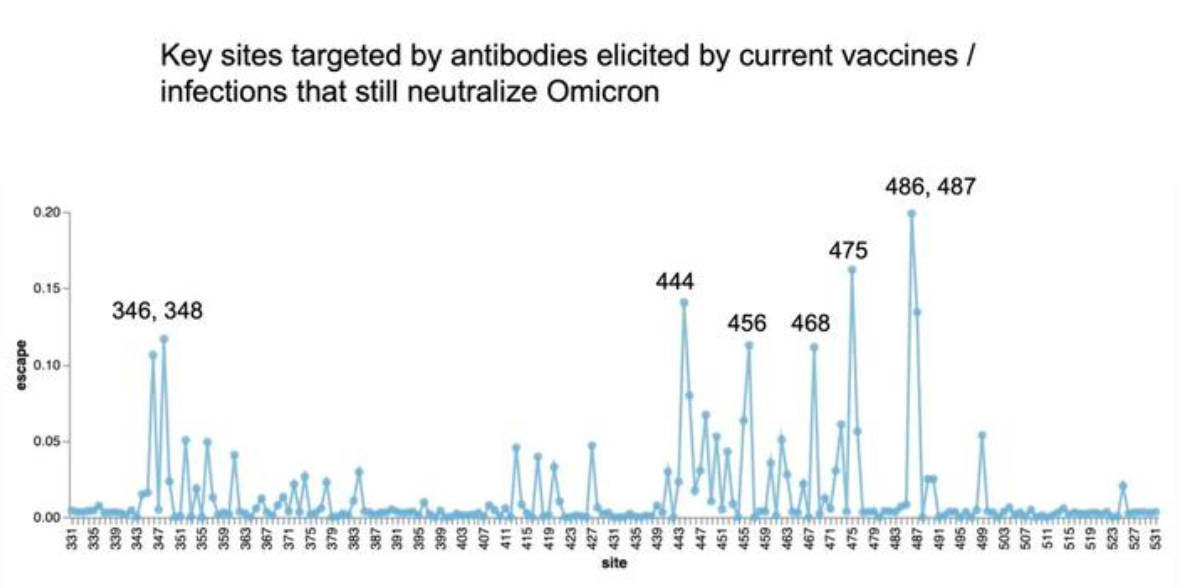
- RBD 区域突变,或者带有可能导致强免疫逃逸或者导致 hACE2 结合力上升的突变,即有流行潜力。 要关注到这里的内容可能就得读一读文献了。例如比较有名的在早期 Bloom Lab 做了突变位点对免疫逃逸能力的关系的研究[3],如下图,也说明了 346, 444, 486 等位点的突变可能会导致免疫逃逸能力大幅增强。(虽然在 XBB 时代这些位点基本上已经全突变完了……
- 要看比较新的结果的话可能就得平时关注关注最新的研究成果,例如曹云龙组最新的论文预测了一组在 XBB.1.5 基础上的图片可能造成的免疫逃逸的结果[4],如下图。图里列出的突变可能就需要重点关注。
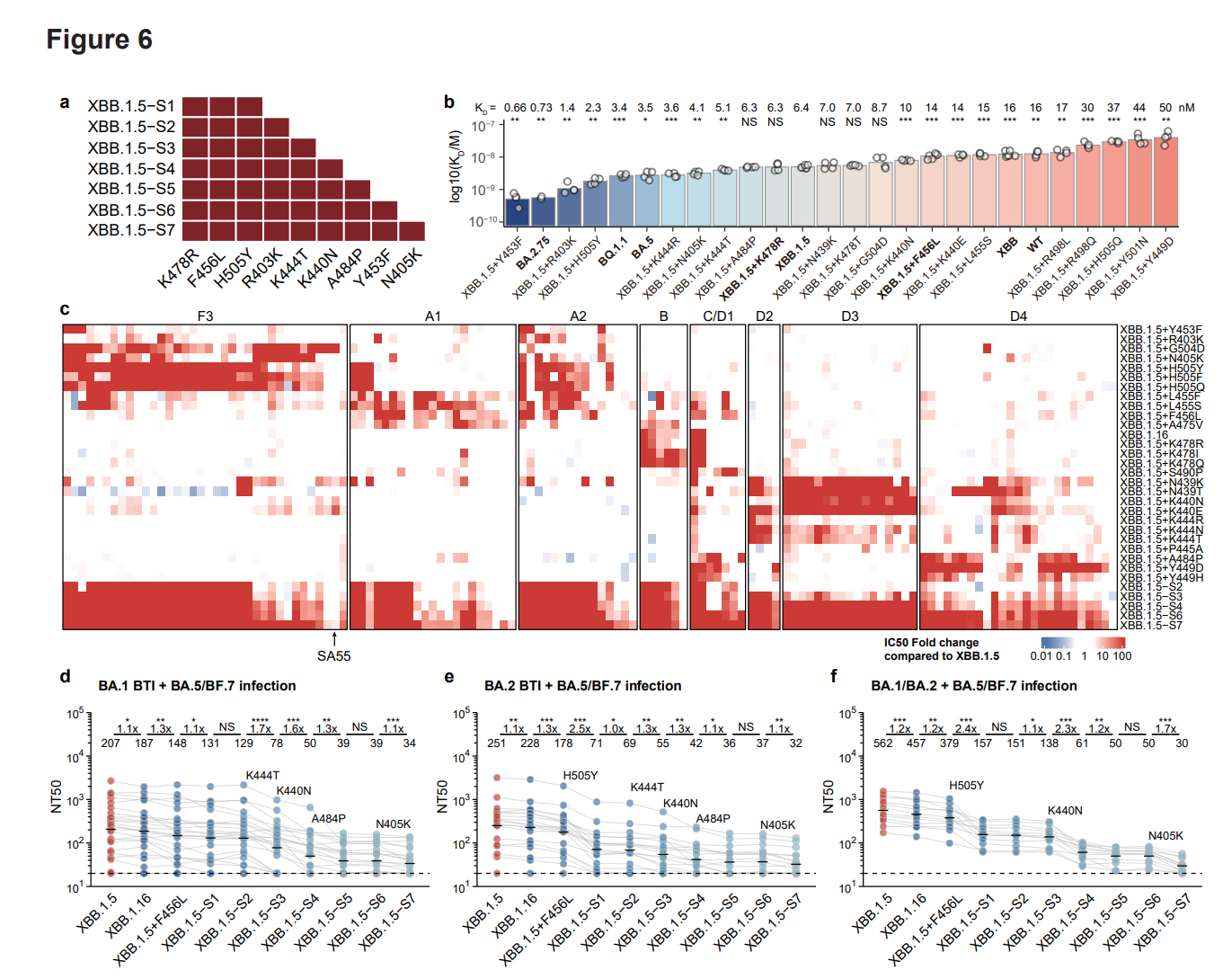
至于在这些范围外的一些突变,可以试试直接搜突变相关的论文,或许有些突变的效果有人研究过。也可以在 Issue 里搜一搜其他毒株是否有相同突变,或许有人贴过参考文献。
能力和篇幅优先,这里无法详细展开,只能大致讲了几个通常会关注的方面。综合这些因素,可以大致评估一个新毒株的重要性。
作为本文例子发现的这一小撮毒株,仅仅是符合新毒株的定义罢了,并没有什么出奇的地方,目前来看还没有获得命名的资格。PANGO 委员会实际上没几个人,大家也别给他们塞垃圾了。
结语
本文只是作为一个业余爱好者简单描述了一下一个新的 SARS-Cov-2 毒株是怎样被发现和命名的,文中举出的例子也是一个非常简单的小例子。在更真实的情况中,可能会遇到更复杂的情形,例如需要费一些力气才能找到合适的 query 区分出一个新的毒株。文中也完全没有提到重组毒株该如何辨别和寻找切割位点,因为这确实更为复杂。文字的一些专业知识,也可能存在错误,请不吝指出。
尽管 COVID-19 正在向 endemic 发展,但各国都仍在监视其变异情况,而这正是各位 Variant Hunter 正在做的事情,这也是一种,社区的力量。
参见
- https://github.com/neherlab/SARS-CoV-2_variant-reports/blob/bacd74f2308749c47d4a47b89115fce26e602d9f/reports/variant_report_2022-09-29.md
- https://twitter.com/JPWeiland/status/1577070487842476032
- https://www.biorxiv.org/content/10.1101/2021.12.04.471236v1
- https://www.biorxiv.org/content/10.1101/2023.05.01.538516v2